Research
Developing high resolution spatial omics technologies to examine the cellular functions of biomedical condensates
Biomolecular condensates are diverse and abundant membrane-less organelles that partition the intracellular environment into functionally distinct compartments containing specific sets of molecules and reactions. Biomolecular condensates participate in various essential housekeeping, stress-response, or cell type-specific processes. Furthermore, an increasing number of studies have linked the physiological state of a cell or the spatial locations of condensates in a cell with the composition of the condensates. Thus, to understand the functions of biomolecular condensates, it is important to know how the molecular content in the condensates change in health vs. disease or in different spatial locations.
To this end, we have been working to develop novel spatial omics technologies that enable multi-modal molecular characterization of biomolecular condensates at the sub-micron scale. These technologies combine high resolution imaging with high throughput sequencing to achieve spatial transcriptome, epigenome, and genome profiling at subcellular resolution.
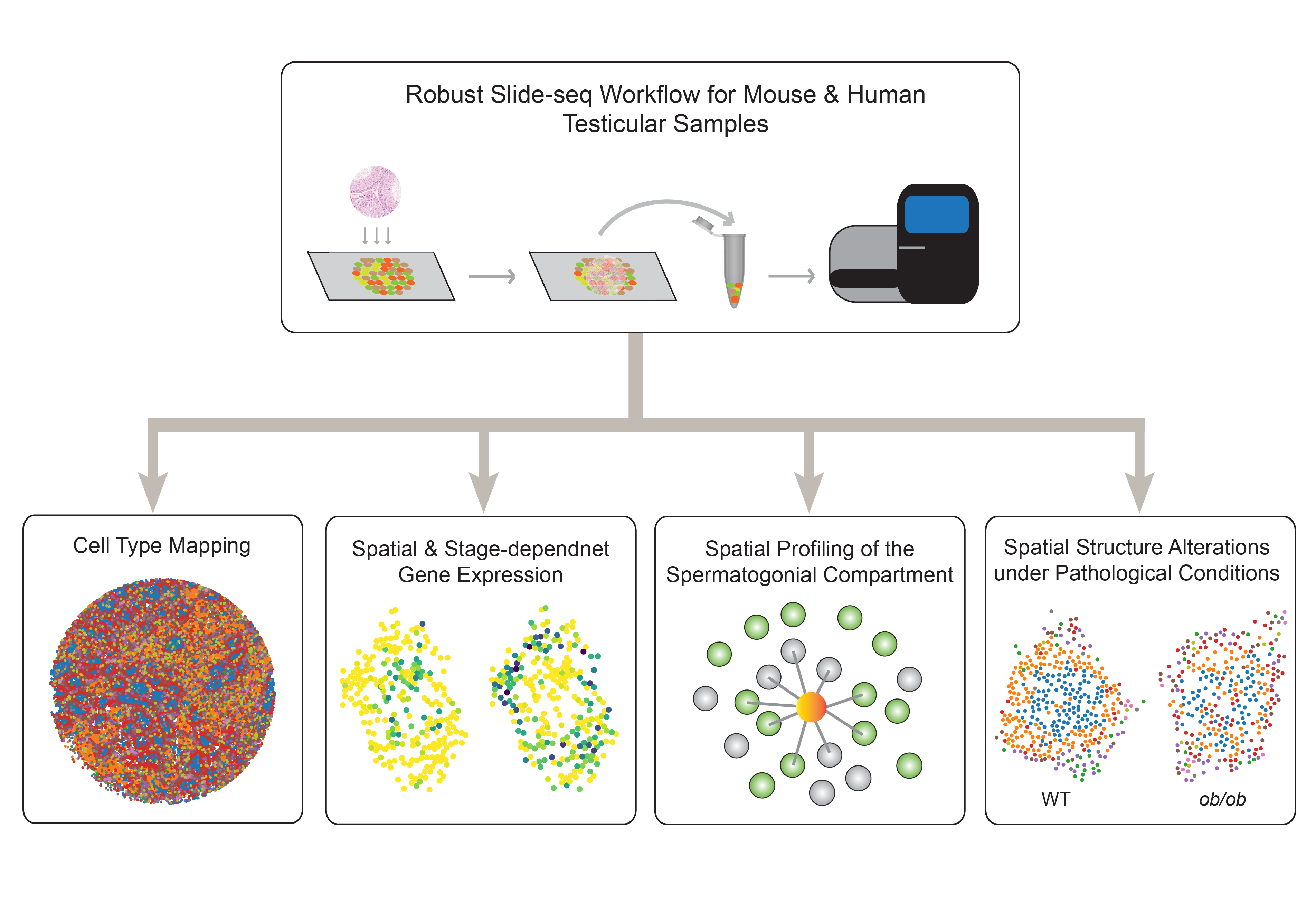
Studying mammalian reproduction through a genomics lens
We are interested in solving the biggest mysteries in mammalian reproduction by leveraging cutting-edge genomics technologies. Some of the curiosity-driven research projects include:
1) How can we achieve a complete human in vitro spermatogenesis?
2) What is the molecular mechanism of immune tolerance of sperm within the female reproductive tract?
3) What are the functions of extrachromosomal circular DNA in the germline?
4) Why and how can some sperm outcompete other sperm within the same ejaculate for fertilization?
5) Why does the testis express the largest number of genes in the body?
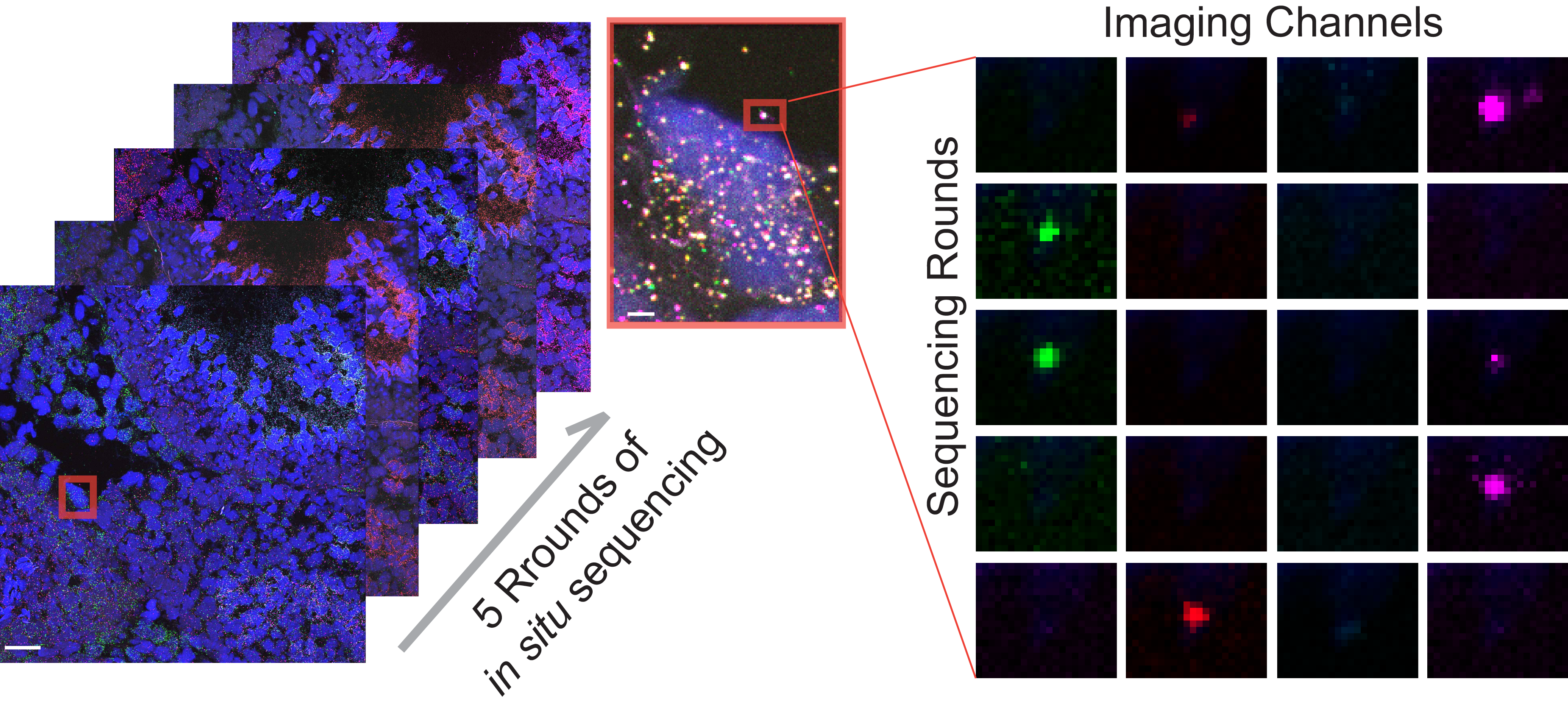
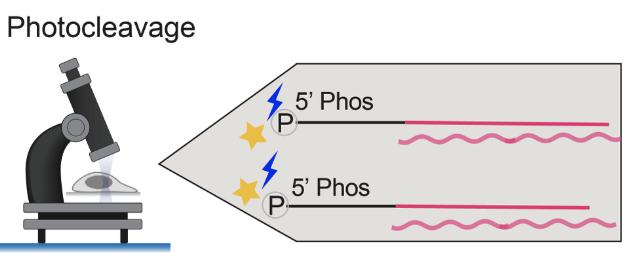
Spatially-resolved functional genomics to study gene functions in the native tissue context
We are developing a high throughput in vivo perturbation screen approach to examine gene functions at scale. To achieve this, we are developing an in situ molecular sequencing platform capable of reading out the identity of each perturbation in the native tissue context. We are using this platform to investigate the functional impacts of genetic perturbations on spermatogensis at an unprecendented scale.